1. Approximately 8% of Australia’s 250,000 annual births occur preterm (prior to 37 weeks completed gestation). Preterm infants represent 75% of all neonatal deaths in Australia, with the vast majority of these deaths due to pulmonary disease.
2. The respiratory consequences for survivors of preterm birth include the immediate challenges of breathing with underdeveloped lungs, usually manifest as respiratory distress syndrome (RDS), and, in the long-term, with persisting pulmonary abnormalities. Therapies to prevent neonatal lung disease now permit survival of preterm infants born as early as 22 weeks’ gestational age, but not without consequences.
3. Preterm infants are at risk of developing chronic lung disease/bronchopulmonary dysplasia (BPD). The lungs of infants dying from BPD are inflamed and have fewer, larger alveoli than normal, and abnormal pulmonary vascular development. There now is a growing appreciation of the contribution of intrauterine inflammation to the aetiology of BPD.
4. Impaired airway function is commonly reported in follow-up studies of children born preterm. Decreased expiratory flow rates have been associated with preterm birth per se but airway function appears more affected in survivors of RDS and BPD. Observations in survivors of BPD suggest persisting abnormalities in the structure of the lung parenchyma and airways.
5. Follow-up studies of preterm infants into adulthood are lacking, as are experimental examinations of the long-term physiological and anatomical effects of preterm birth. Both are necessary to understand the causes of the long-term respiratory consequences of preterm birth.
Preterm birth is defined as birth before 37 completed weeks of gestation, and can be further defined according to severity: very preterm births are those before 34 weeks’ gestation and extremely preterm births occur before 27 weeks.1 Delivery before 22 completed weeks of gestation is usually fatal but there are reports of infants who have survived birth after 21 weeks and 5 days of gestation.2
In Australia, preterm birth occurs in 7.9% of pregnancies (rates for individual states vary from 7.1-11%), representing 20,000 of the country’s 250,000 annual births.3 The incidence of preterm birth is similar in most developed countries. In recent years there has been a trend for preterm birth rates to increase.1 This is likely due, in part, to increased rates of intervention; but the fact that rates of very preterm and extremely preterm births are increasing demonstrates that changes in clinical management are not the sole cause.
In 1944, Eastman recognised that, “only when the factors causing prematurity are clearly understood, can any intelligent attempt at prevention be made”. Our failure to predict and prevent preterm births, despite a great deal of effort in the intervening decades, reflects the complex and varied causes.
There are a number of factors associated with preterm birth: multiple gestation, previous preterm birth, maternal/fetal complications, low socio-economic status, drug use and assisted reproduction all increase the risk of preterm birth. The mechanisms responsible for these well-established associations are incompletely understood. In recent years, elucidation of at least some of the mechanisms of preterm birth has appeared promising as a result of the growing recognition that intrauterine infection or inflammation is common in cases of preterm birth.4
The cost of preterm birth is immense. Data from the USA indicate that babies born preterm (9% of live births) account for $5.8 billion (57%) of the total $10.2 billion annual cost of neonatal care in that country.5 Costs for individual infants vary according to gestational age (it costs approximately $250,000 to support a single infant born at 25 weeks) but the rates of preterm birth vary in a reciprocal fashion, such that (in the USA) total costs are $38,000,000 for all deliveries in each gestational week age group from 25 to 36 weeks.6 The high costs of support for extremely preterm and very preterm infants parallel their requirements for ventilatory support.6
Respiratory disease is the single greatest cause of illness and death in preterm infants. The immediate respiratory consequences of preterm birth can result from pulmonary prematurity per se, abnormalities in prenatal lung development arising from factors associated with preterm birth, and the medical management of the preterm infant.
Structural and biochemical maturation of the fetal lung does not normally occur until late in gestation. Surfactant deficiency and structural immaturity of the preterm lung both contribute to neonatal respiratory distress syndrome (RDS); this is reflected by the association between gestational age at delivery and the incidence of RDS.6 Symptoms of RDS include tachypnea, chest wall retraction and cyanosis, and the chest has a ‘ground glass’ appearance on X-Ray.
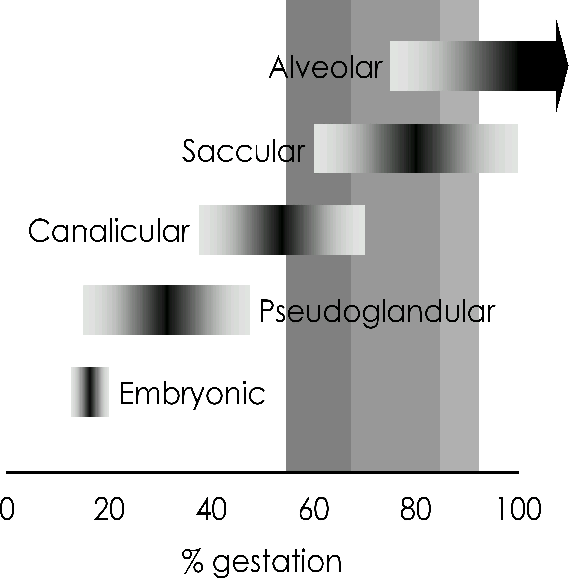
Extremely preterm birth corresponds with the canalicular stage of lung development (see Figure), during which vascularisation of the dense mesenchyme is only beginning and there is little differentiation of airway epithelial cells into types I (across which gas exchange occurs) and II (the surfactant-producing cells). Very preterm birth coincides with the saccular stage of lung development; the pulmonary blood/air barrier has only commenced thinning at this stage, vascularisation is incomplete, and type II epithelial cells are immature.7 Neither of these developmental stages is consistent with independent ventilation and the vast majority of infants born at these ages require ventilatory support.6
Figure 1. Stages of structural lung development. Shaded vertical bars indicate periods of gestation corresponding to preterm birth (light shading), very preterm birth (medium shading) or extremely preterm birth (dark shading). Adapted from Pringle, 1986.7
Two strategies used in perinatal medicine have contributed drastically to reducing the incidence and impact of RDS. Administration of antenatal corticosteroids to women at risk of preterm delivery reduces the risk of RDS in their offspring by ∼50%,8 and surfactant administration to preterm neonates reduces the severity of RDS, the progression of respiratory disease, and the incidence of death9-11.
Evidence from experimental studies in animals indicates that corticosteroid-induced functional ‘maturation’ of the preterm lungs is due primarily to structural changes and that increases in surfactant are relatively slower to occur and exist only transiently after corticosteroid treatment.12 Thus, surfactant deficiency (the principal contributor to RDS) may not be adequately treated by antenatal corticosteroids; the fact that RDS is prevented by antenatal corticosteroids in only 50% of cases reflects this.
The structural changes induced by antenatal corticosteroid treatment result in thinner alveolar walls and fewer, larger alveoli than normal.13 Although beneficial in the immediate neonatal period, these structural effects may have longer-term adverse consequences because this phenotype is characteristic of the chronic neonatal lung disease, bronchopulmonary dysplasia (BPD). Meta-analysis of available data from 3 randomised controlled trials certainly demonstrates that antenatal corticosteroids do not reduce the risk of chronic lung disease.8 The ideal strategy for inducing preterm lung maturation remains to be discovered.
The recent realisation that many preterm births are associated with intrauterine inflammation, and that intrauterine infection/inflammation is associated with a reduced risk of RDS in some cohorts of infants14,15, prompted us to examine the effects of intrauterine inflammation on lung maturation in sheep. Using a model of chorioamnionitis, based on intra-amniotic administration of lipopolysaccharide (LPS) from E. coli, we have shown that fetal lung inflammation causes striking improvements in preterm lung function,16 due primarily to increases in pulmonary surfactant17 (remodelling of the lung parenchyma also occurs). Lung ‘maturation’ induced by intra-amniotic LPS is likely due to locally-produced mediators of the inflammatory response18 acting directly on the lungs,19 rather than by increases in endogenous corticosteroids.20,21 Intra-amniotic injection of the pro-inflammatory cytokine interleukin-1 (IL-1) has similar inflammatory and ‘maturational’ effects on the preterm lungs to intra-amniotic LPS22 but tumour necrosis factor alpha (TNF-α) does not.23 Activation of distinct ‘maturational’ pathways by inflammation and corticosteroids is suggested by augmentation of responses when treatments are combined;24 however, observational data from humans suggests that the reduced risk of RDS associated with chorioamnionitis is not further reduced by corticosteroid treatment.25 Exploitation of inflammatory pathways for inducing preterm lung ‘maturation’ in humans appears a distant possibility; the reduced risk of RDS associated with inflammation is accompanied by an increased risk of BPD in humans,15 and we have observed BPD-like changes in lung structure and the surfactant system in preterm lambs after exposure to intrauterine inflammation.26
Bronchopulmonary dysplasia was first described in 1967 by Northway et al.,27 as a form of chronic lung disease of preterm infants, attributable to the injurious effects of high oxygen levels and airway pressures used during mechanical ventilation.27 Experimental studies using preterm baboons established the role of these factors in the aetiology and pathology of BPD.28 This classical form of BPD is rare now, because of the use of antenatal corticosteroids, surfactant and gentle ventilation strategies. Instead, a ‘new BPD’, defined as the requirement for supplemental oxygen at 36 weeks’ post-conceptional age,29 has emerged; affecting the most premature of infants. These infants may have very mild or no respiratory disease initially, but develop increasing needs for ventilatory support; pathological investigation in fatal cases reveals lungs with fewer, larger alveoli than controls and impaired vascular development.29 This ‘new BPD’ phenotype is observed in ventilated very premature baboons, despite the use of surfactant and relatively gentle ventilation.30
It is becoming increasingly evident that BPD is often associated with pulmonary inflammation originating during fetal life.29,31 Tracheal colonisation by micro-organisms at the time of birth, reflecting prenatal colonisation, is associated with an increased risk of BPD14,32 and, as mentioned above, our experiments in sheep demonstrate BPD-like changes in surfactant and lung structure after exposure to intra-amniotic LPS.26 Preterm baboons delivered after intra-amniotic injection of Ureaplasma urealyticum, and ventilated for 14 days, had impaired lung function and more severe histopathology than controls, demonstrating that prenatal infection worsens postnatal respiratory outcomes.33
The pathological features of BPD suggest that structural development of the lung has been arrested at an immature stage, and that normal septation and alveolarisation has been inhibited.34 In the neonatal lung, alveolarisation can be inhibited by inflammation, corticosteroids, and hyperoxia;35 each of these is a consequence of standard management for many preterm neonates. Prenatal insults can also inhibit alveolarisation; maternal corticosteroid treatment and intra-amniotic LPS injection both result in fewer, larger alveoli than normal, in the lungs of preterm lambs.36 Interestingly, the fetal lungs appear to have a capacity to normalise their development after a period of inhibition of alveolarisation if delivery does not occur until late in gestation.13,37 Unfortunately, normalisation of lung structure after preterm birth is more elusive.
In recent years, a variety of ventilatory strategies have been developed for the management of preterm infants, aimed at reducing lung injury and avoiding BPD. Studies of preterm baboons have shown that high frequency oscillatory ventilation (HFOV) results in better respiratory function and less inflammation than gentle positive pressure ventilation (with low oxygen levels and volumes), but inhibition of alveolarisation occurred using both strategies.38 Clinical data are consistent with these findings; meta-analysis of randomised controlled trials has shown that HFOV does not reduce the risk of BPD.39
Comparison of rates of BPD in different intensive care units in North America in the 1980s showed that the use of continuous positive airway pressure (CPAP) at Columbia University was associated with the best outcomes for preterm infants and the lowest incidence of BPD.40 This observation led to a number of recent experimental and clinical investigations of CPAP for support of preterm neonates. Our own investigations have shown that there is less initial inflammation in the lungs of lambs supported from birth by CPAP than in mechanically-ventilated lambs.41 Recent studies of CPAP support for preterm baboons have demonstrated that its use is associated with avoidance of significant lung disease over the first postnatal month.42 Perhaps most importantly, these studies showed that lung structure in 28 day-old baboons, delivered very preterm and supported with CPAP, was comparable with that of a group of unventilated late-gestation fetal baboons.42 These results demonstrate that preservation of normal lung development in preterm infants is a possibility and provide encouraging results for the more routine application of CPAP.
In addition to surfactant deficiency and pulmonary structural immaturity (and/or abnormalities) associated with preterm birth, the composition and function of surfactant that is present in the lungs of preterm infants may be abnormal. The levels of surfactant protein (SP)-A, SP-B and SP-C in tracheal aspirate samples collected from 2-day-old preterm neonates are only a fraction of concentrations in preterm infants older than 1 week, which are comparable to adult levels.43 In preterm infants with deteriorating respiratory status, levels of SP-A, SP-B and SP-C are reduced and surfactant function is compromised.44 While administration of exogenous surfactant improves outcomes for preterm infants, surfactant function is not normalised by synthetic surfactants.45 Exogenous surfactants extracted from animal lungs contain SP-B and SP-C, which are important for conferring the surface-active properties of surfactant, and these preparations appear superior to synthetic versions that lack surfactant proteins.46
There have been a number of studies examining lung function in children born preterm but there is little information on the consequences in adulthood of preterm birth. The general impression from studies of children is that those born preterm have higher rates of respiratory illness than those born at term;47,48 respiratory flow rates are lower,49,50 diffusing capacity may be reduced,50,51 and airway reactivity may be increased.48,49 These adverse effects of preterm birth appear to be exacerbated by RDS52 or BPD48,51,53. BPD in infancy is also associated with airway obstruction and reactivity in adolescence/early adulthood.54 These adverse respiratory consequences of preterm birth are likely the consequence of persisting pulmonary structural abnormalities.
Normal lung development requires a series of carefully orchestrated events that can be compromised by a variety of prenatal and postnatal factors associated with preterm birth. Given the substantial financial cost of respiratory illness in preterm infants, and the likely life-long burden of respiratory disease associated with preterm birth, there is a critical need to identify the mechanisms by which lung development is altered in preterm infants, and to develop management strategies for preterm labour, and means of providing support to preterm infants, that have minimal impact on lung growth.
1. Tucker J, McGuire W. Epidemiology of preterm birth. BMJ 2004; 329: 675-8.
2. Guinness World Records. 2005. http://www.guinnessworldrecords.com
3. Laws P, Sullivan E, Australia's mothers and babies 2000, in Perinatal Statistics Series Number 15, AIHW National Perinatal Statistics Unit, Editor. 2004, Australian Institute of Health and Welfare: Canberra.
4. Goldenberg RL, Hauth JC, Andrews WW. Intrauterine infection and preterm delivery. N. Engl. J. Med. 2000; 342: 1500-7.
5. St John EB, Nelson KG, Cliver S P, Bishnoi RR, Goldenberg RL. Cost of neonatal care according to gestational age at birth and survival status. Am. J. Obstet. Gynecol. 2000; 182: 170-5.
6. Gilbert WM, Nesbitt TS, Danielsen B. The cost of prematurity: quantification by gestational age and birth weight. Obstet. Gynecol. 2003; 102: 488-92.
7. Pringle KC. Human fetal lung development and related animal models. Clin. Obstet. Gynecol. 1986; 29: 502-13.
8. Crowley P. Prophylactic corticosteroids for preterm birth. Cochrane Database Syst. Rev. 1996.
9. Soll R. Prophylactic synthetic surfactant for preventing morbidity and mortality in preterm infants. Cochrane Database Syst. Rev. 1997.
10. Soll R, Morley C. Prophylactic versus selective use of surfactant in preventing morbidity and mortality in preterm infants. Cochrane Database Syst Rev. 2001;(2):CD000510
11. Soll R. Prophylactic natural surfactant extract for preventing morbidity and mortality in preterm infants. Cochrane Database Syst. Rev. 1998.
12. Jobe AH, Ikegami M. Lung development and function in preterm infants in the surfactant treatment era. Ann. Rev. Physiol. 2000; 62: 825-46.
13. Willet KE, Jobe AH, Ikegami M, Kovar J, Sly PD. Lung morphometry after repetitive antenatal glucocorticoid treatment in preterm sheep. Am. J. Respir. Crit. Care Med. 2001; 163: 1437-43.
14. Hannaford K, Todd DA, Jeffery H, John E, Blyth K, Gilbert GL. Role of ureaplasma urealyticum in lung disease of prematurity. Arch. Dis. Child Fetal Neonatal Ed. 1999; 81: F162-7.
15. Watterberg KL, Demers LM, Scott SM, Murphy S. Chorioamnionitis and early lung inflammation in infants in whom bronchopulmonary dysplasia develops. Pediatrics 1996; 97: 210-5.
16. Jobe AH, Newnham JP, Willet KE, et al. Effects of antenatal endotoxin and glucocorticoids on the lungs of preterm lambs. Am. J. Obstet. Gynecol. 2000; 182: 401-8.
17. Bachurski CJ, Ross GF, Ikegami M, Kramer BW, Jobe AH. Intra-amniotic endotoxin increases pulmonary surfactant proteins and induces SP-B processing in fetal sheep. Am. J. Physiol. Lung Cell Mol. Physiol. 2001; 280: L279-85.
18. Kramer BW, Moss TJ, Willet KE, et al. Dose and time response after intraamniotic endotoxin in preterm lambs. Am. J. Respir. Crit. Care Med. 2001; 164: 982-8.
19. Moss TJ, Nitsos I, Kramer BW, Ikegami M, Newnham JP, Jobe AH. Intra-amniotic endotoxin induces lung maturation by direct effects on the developing respiratory tract in preterm sheep. Am. J. Obstet. Gynecol. 2002; 187: 1059-65.
20. Nitsos I, Moss TJ, Cock ML, Harding R, Newnham JP. Fetal responses to intra-amniotic endotoxin in sheep. J. Soc. Gynecol. Investig. 2002; 9: 80-5.
21. Jobe AH, Newnham JP, Willet KE, et al. Endotoxin-induced lung maturation in preterm lambs is not mediated by cortisol. Am. J. Respir. Crit. Care Med. 2000; 162: 1656-61.
22. Willet KE, Kramer BW, Kallapur SG, et al. Intra-amniotic injection of IL-1 induces inflammation and maturation in fetal sheep lung. Am. J. Physiol. Lung Cell Mol. Physiol. 2002; 282: L411-20.
23. Ikegami M, Moss TJ, Kallapur SG, et al. Minimal lung and systemic responses to TNF-alpha in preterm sheep. Am. J. Physiol. Lung Cell Mol. Physiol. 2003; 285: L121-9.
24. Newnham JP, Moss TJ, Padbury J F, et al. The interactive effects of endotoxin with prenatal glucocorticoids on short-term lung function in sheep. Am. J. Obstet. Gynecol. 2001; 185: 190-7.
25. Foix-L'helias L, Baud O, Lenclen R, Kaminski M, Lacaze-Masmonteil T. Benefit of antenatal glucocorticoids according to the cause of very premature birth. Arch. Dis. Child Fetal Neonatal Ed. 2005; 90: F46-8.
26. Moss TJ, Newnham JP, Willett K E, Kramer BW, Jobe AH, Ikegami M. Early gestational intra-amniotic endotoxin: lung function, surfactant, and morphometry. Am. J. Respir. Crit. Care Med. 2002; 165: 805-11.
27. Northway WH, Jr., Rosan RC, Porter DY. Pulmonary disease following respirator therapy of hyaline-membrane disease. Bronchopulmonary dysplasia. N. Engl. J. Med. 1967; 276: 357-68.
28. Coalson JJ, Kuehl TJ, Escobedo M B, et al. A baboon model of bronchopulmonary dysplasia. II. Pathologic features. Exp. Mol. Pathol. 1982; 37: 335-50.
29. Jobe AH, Bancalari E. Bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 2001; 163: 1723-9.
30. Coalson JJ, Winter VT, Siler-Khodr T, Yoder BA. Neonatal chronic lung disease in extremely immature baboons. Am. J. Respir. Crit. Care Med. 1999; 160: 1333-46.
31. Speer CP. Inflammation and bronchopulmonary dysplasia. Semin. Neonatol. 2003; 8: 29-38.
32. Young KC, Del Moral T, Claure N, Vanbuskirk S, Bancalari E. The Association between early tracheal colonization and bronchopulmonary dysplasia. J. Perinatol. 2005; 25: 403-7.
33. Yoder BA, Coalson JJ, Winter V T, Siler-Khodr T, Duffy LB, Cassell GH. Effects of antenatal colonization with ureaplasma urealyticum on pulmonary disease in the immature baboon. Pediatr. Res. 2003; 54: 797-807.
34. Jobe AJ. The new BPD: an arrest of lung development. Pediatr. Res. 1999; 46: 641-3.
35. Jobe AH. Glucocorticoids, inflammation and the perinatal lung. Semin. Neonatol. 2001; 6: 331-42.
36. Willet KE, Jobe AH, Ikegami M, Newnham J, Brennan S, Sly PD. Antenatal endotoxin and glucocorticoid effects on lung morphometry in preterm lambs. Pediatr. Res. 2000; 48: 782-8.
37. Kallapur SG, Nitsos I, Moss TJ, et al. Chronic endotoxin exposure does not cause sustained structural abnormalities in the fetal sheep lungs. Am. J. Physiol. Lung Cell Mol. Physiol. 2005; 288: L966-74.
38. Yoder BA, Siler-Khodr T, Winter V T, Coalson JJ. High-frequency oscillatory ventilation: effects on lung function, mechanics, and airway cytokines in the immature baboon model for neonatal chronic lung disease. Am. J. Respir. Crit. Care Med. 2000; 162: 1867-76.
39. Henderson-Smart DJ, Bhuta T, Cools F, Offringa M. Elective high frequency oscillatory ventilation versus conventional ventilation for acute pulmonary dysfunction in preterm infants. Cochrane Database Syst Rev. 2003: (4) CD000104.
40. Avery ME, Tooley WH, Keller JB, et al. Is chronic lung disease in low birth weight infants preventable? A survey of eight centers. Pediatrics 1987; 79: 26-30.
41. Jobe AH, Kramer BW, Moss TJ, Newnham JP, Ikegami M. Decreased indicators of lung injury with continuous positive expiratory pressure in preterm lambs. Pediatr. Res. 2002; 52: 387-92.
42. Thomson MA, Yoder BA, Winter V T, et al. Treatment of immature baboons for 28 days with early nasal continuous positive airway pressure. Am. J. Respir. Crit. Care Med. 2004; 169: 1054-62.
43. Ballard PL, Merrill JD, Godinez RI, Godinez MH, Truog WE, Ballard RA. Surfactant protein profile of pulmonary surfactant in premature infants. Am. J. Respir. Crit. Care Med. 2003; 168: 1123-8.
44. Merrill JD, Ballard RA, Cnaan A, et al. Dysfunction of pulmonary surfactant in chronically ventilated premature infants. Pediatr. Res. 2004; 56: 918-26.
45. McMillan DD, Singhal N, Shukla A K, Schurch S. Tracheal aspirate surface tension in babies with hyaline membrane disease: effects of synthetic surfactant replacement. Pediatr. Pulmonol. 1998; 26: 173-82.
46. Ainsworth SB, Beresford MW, Milligan DW, et al. Pumactant and poractant alfa for treatment of respiratory distress syndrome in neonates born at 25-29 weeks' gestation: a randomised trial. Lancet. 2000; 355: 1387-92.
47. Rona RJ, Gulliford MC, Chinn S. Effects of prematurity and intrauterine growth on respiratory health and lung function in childhood. BMJ 1993; 306: 817-20.
48. Gross SJ, Iannuzzi DM, Kveselis DA, Anbar RD. Effect of preterm birth on pulmonary function at school age: a prospective controlled study. J. Pediatr. 1998; 133: 188-92.
49. Pelkonen AS, Hakulinen AL, Turpeinen M. Bronchial lability and responsiveness in school children born very preterm. Am. J. Respir. Crit. Care Med. 1997; 156: 1178-84.
50. Hakulinen AL, Jarvenpaa AL, Turpeinen M, Sovijarvi A. Diffusing capacity of the lung in school-aged children born very preterm, with and without bronchopulmonary dysplasia. Pediatr. Pulmonol. 1996; 21: 353-60.
51. Mitchell SH, Teague WG. Reduced gas transfer at rest and during exercise in school-age survivors of bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 1998; 157: 1406-12.
52. Cano A, Payo F. Lung function and airway responsiveness in children and adolescents after hyaline membrane disease: a matched cohort study. Eur. Respir. J. 1997; 10: 880-5.
53. Kilbride HW, Gelatt MC, Sabath R J. Pulmonary function and exercise capacity for ELBW survivors in preadolescence: effect of neonatal chronic lung disease. J. Pediatr. 2003; 143: 488-93.
54. Northway WH, Jr., Moss RB, Carlisle KB, et al. Late pulmonary sequelae of bronchopulmonary dysplasia. N. Engl. J. Med. 1990; 323: 1793-9.